- Visibility 181 Views
- Downloads 46 Downloads
- Permissions
- DOI 10.18231/j.ijpo.2024.088
-
CrossMark

Myxoid glioneuronal tumor of septum pellucidum: A rare case report and review of literature
- Author Details:
-
 Krupa Mehta *
Krupa Mehta *
-
Riddhi Parsana
-
Raj Shah
-
Devanshi Shah
-
Preeti Jhaveri
-
Minesh Gandhi
-
Cherry Shah
Abstract
Myxoid glioneuronal tumours (MGNTs) represent a recently recognized entity in the World Health Organization (WHO) classification of Central Nervous System (CNS) tumours 5th edition. They are often seen in specific locations within the CNS, such as the septum pellucidum, foramen of Monro, or periventricular white matter of the lateral ventricle. Historically, these tumours were frequently misclassified as dysembryoplastic Neuroepithelial Tumours (DNT), primarily due to their similar histological features. MGNTs have been related with a dinucleotide substitution at codon 385 within the platelet-derived growth factor receptor alpha (PDGFRA) gene. This genetic variation results in the replacement of a lysine residue with either leucine or isoleucine (p. LysK385Leu/Iso). This genetic alteration appears to be unique to MGNTs and has not been reported in any other CNS tumor types.
Introduction
Myxoid glioneuronal tumor (MGNT) is a benign glioneuronal neoplasm recently introduced in the 5th edition of the World Health Organization (WHO) classification of the central nervous system (CNS) tumours 2021. Until now only few cases have been reported. MGNT represents about 2% of all brain tumor. It predominantly affects children and young adult with the highest incidence in second and third decade of life. Radiographically, MGNT have a preference for origin in the septum pellucidum but have also been observed in the genu of the corpus callosum and periventricular white matter of the lateral ventricle. Previously, these tumours were often misdiagnosed or classified under the umbrella of dysembryoplastic neuroepithelial tumours (DNTs), due to their histological similarities with classical cortical DNTs. MGNTs have been related with a dinucleotide substitution at codon 385 within the platelet-derived growth factor receptor alpha (PDGFRA) gene. This genetic variation results in the replacement of a lysine residue with either leucine or isoleucine (p. LysK385Leu/Iso). This genetic variation has not been reported in any other CNS tumor.
The clinical diagnosis of a myxoid glioneuronal tumor involves a combination of clinical presentation, radiological imaging, and histopathological evaluation. The most common clinical presentation includes headaches, emesis, seizures, and behavioural disturbances, although the presenting symptoms may vary. On neuroimaging (Computed Tomography Scan), these tumours show well-circumscribed margins, cystic features, and a discrete size (mostly 10 to 30 mm). On Magnetic Resonance Imaging, they show mostly low intensity on T1 weighted imaging and high intensity on T2 weighted imaging. There is no contrast enhancement or restricted diffusion. Calcifications and multinodularity, which are common in cortical DNTs, but not seen in MGNT. MGNTs are mostly presented with proliferation of oligodendrocyte- like cells lodged in a prominent myxoid stroma. Histologic features are reminiscent of either DNT or rosette-forming glioneuronal tumor (RGNT).
Case Presentation
A 51 years old male patient with average built and height came to Neurosurgery department with chief complains of forgetfulness for 1 month, bowel and bladder incontinence for 1 months and imbalance while walking in the last 3 days. Patient was advised MRI brain with cerebral angiography.
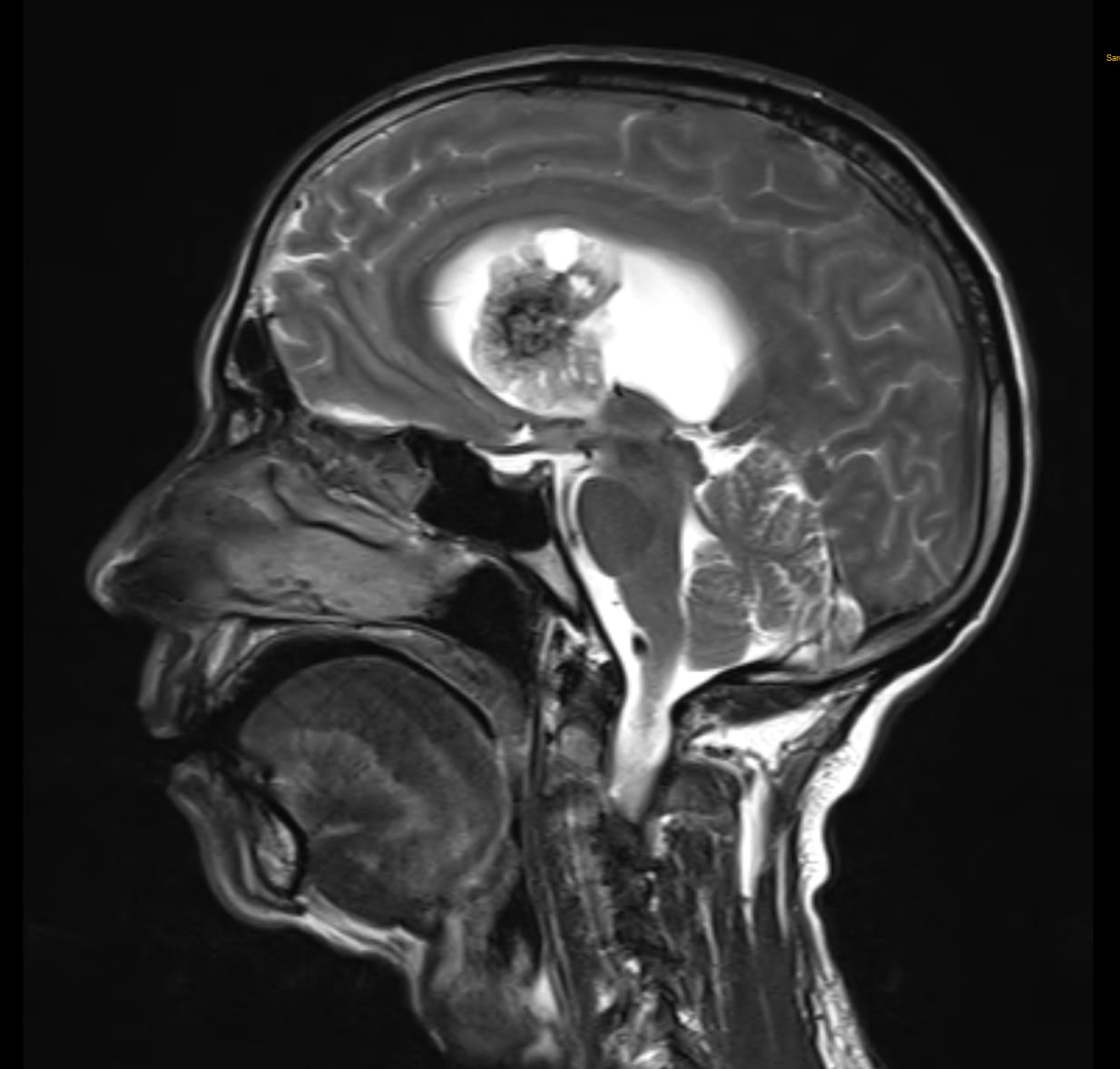
On follow up visit: MRI brain with cerebral angiography findings showed isointense on T1W imaging and hypointense on T2WI / FLAIR. Well-defined heterogeneously enhancing lobulated altered signal intensity lesion with few internal haemorrhagic and cystic areas arising from septum pellucidum and cause its mild displacement towards left side suggestive of neoplastic etiology- Central Neurocytoma.
MRI brain with contrast study showed similar findings as MRI angiography. As per MRI report wide excision of supratentorial SOL under general anaesthesia was planned.
All haematological and biochemical investigations like complete blood count, coagulation profile, liver function test and renal function tests were within normal range.
On local examination patient was oriented, conscious and obeyed command.
A wide excision of supratentorial SOL was done under general anaesthesia was sent for histopathological examination.
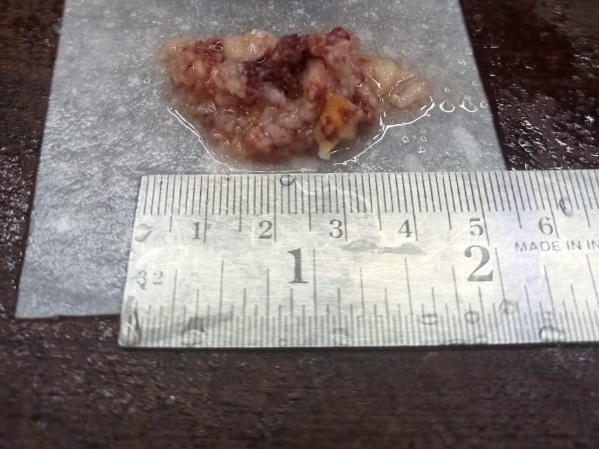
Grossly, specimen consisted of multiple white brown soft tissue portions total measuring 2.5 x 1.5 cm in aggregate.
The microscopy revealed tumor showing neurocystic and glial components. Neurocystic cells are having small round nuclei with fine stippled chromatin and scant cytoplasm. The neurocystic cells are present in partly microcystic arrangement. Glial fibrillary component with oligodendroglia-like cells are seen. Cystic and haemorrhagic areas with hemosiderin laden pigments are seen.

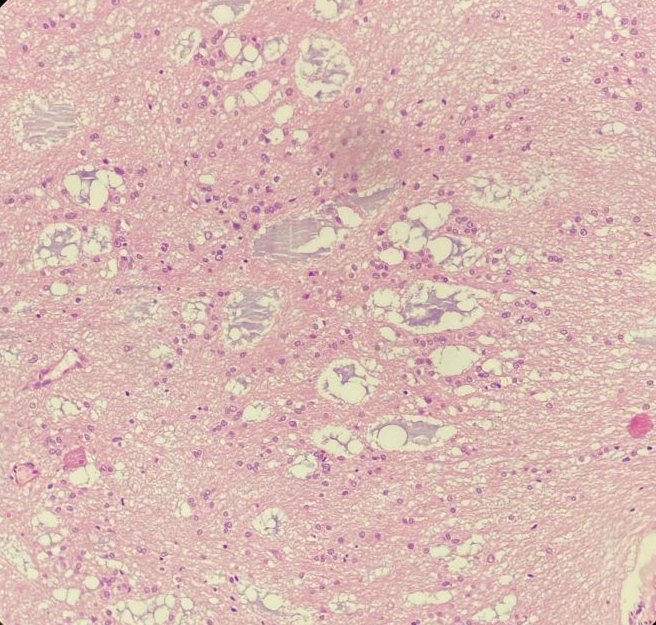
On H&E sections, it was provisionally diagnosed as Low grade Glioneuronal Tumor. Because of its location of septum pellucidum and keeping in mind the differentials, immunohistochemistry (IHC) was advised for confirmation. On IHC, GFAP (Glial fibrillary acidic protein) is positive, SOX 10 is positive and Synaptophysin is negative for oligodendrocyte-like cells and positive for floating neurons confirming our diagnosis of Myxoid Glioneuronal Tumor (WHO Grade 1).




Discussion
MGNT is a well-demarcated tumour of glial neuronal origin that was initially described and reported by Solomon et al in 2018.[1] According to 5th edition of the WHO Classification of Tumours of the Central Nervous System (2021), MGNT has been recognized as “neuronal and mixed neuronal-glial tumours”.[2] MGNT has been assigned Grade 1 according to the WHO CNS classification. Myxoid glioneuronal tumours (MGNTs) are frequently localized to the septum pellucidum, with less common occurrences in the corpus callosum and periventricular white matter of the lateral ventricle.
MGNTs were often previously misdiagnosed as dysembryoplastic neuroepithelial tumours (DNTs) due to their histological similarities, most often located in the septum pellucidum. The histological features of MGNTs can closely resemble those of classical cortical DNTs, as well as rosette-forming glioneuronal tumours (RFGTs), making accurate diagnosis challenging without careful consideration of clinical and molecular characteristics.
MGNT mostly affects children and young adults, with the highest incidence in the second and third decades of life with equal sex distribution.[3] The most common clinical manifestations include headaches, emesis, seizures, and behavioural disturbances, although the presenting symptoms may vary, particularly when these symptoms occur in the context of lesions localized to the septum pellucidum, corpus callosum, or periventricular white matter.[4] It's noteworthy that some patients may be incidentally diagnosed through routine head imaging performed for unrelated reasons. The ability to detect MGNTs incidentally highlights the importance of vigilant radiological assessment and raises the possibility of identifying asymptomatic cases that may benefit from early intervention.
Few cases reported as MGNT arising in a mesencephalic location. Specific considerations and challenges come into play due to the critical functions and complex anatomy of the midbrain. Clinically present as hemiparesis or hemisensory loss, double vision, difficulty with eye movements (due to involvement of cranial nerves III and IV), facial numbness or weakness, ataxia or difficulties with balance and coordination, altered consciousness or behavioural changes.
As per imaging characteristics, MGNT often appears as a rounded or lobulated mass with well-circumscribed borders which is ranging in size from 10 to 30 mm. Computed tomography (CT) scans findings suggest lesions present as hypodense masses. Magnetic resonance imaging (MRI) findings of MGNT suggest more distinctive which typically showing a cystic appearance. These cystic areas always exhibit T1- hypointensity, T2-hyperintensity images. It is particularly high signal intensity on FLAIR sequences (especially around cysts). Most of cases do not demonstrate contrast enhancement or diffusion restriction.[5], [6]
Histologically, MGNTs (Myxoid Glioneuronal Tumours) are predominantly composed of oligodendrocyte-like cells lodged within prominent myxoid stroma. They present histologic characteristics which are reminiscent of Dysembryoplastic Neuroepithelial Tumours (DNTs) or Rosette-forming Glioneuronal Tumours (RGNTs). Previously, these tumours have been misdiagnosed and misclassified as DNT or DNT-like neoplasms of the septum pellucidum. MGNTs are distinct in a way that they do not manifest the BRAF or FGFR1 variants or rearrangements, gene fusions, or kinase domain tandem duplications that are typical in most DNTs and RGNTs.[1]
The oligodendrocyte-like neoplastic cells in Myxoid Glioneuronal Tumors (MGNTs) exhibit a specific immunohistochemical profile. They are positive for OLIG2, SOX10, GFAP, and MAP2, and negative for synaptophysin. The oligodendrocyte-like neoplastic cells in MGNTs are synaptophysin negative, indicating a lack of synaptic vesicles and mature neuronal characteristics while the floating neurons, perivascular neuropil, and neurocystic rosettes are synaptophysin positive, reflecting their mature neuronal features. This differential staining highlights the diverse cellular composition and differentiation states within MGNTs. CD34 staining is restricted to vessels, and the proliferation index is typically low.[7] The Ki-67 labelling index is low (<5%).
As in our case, the lesion was isointense on T1W imaging and hypointense on T2WI / FLAIR. The lesion exhibited well-defined heterogeneously enhancing lobulated altered signal intensity with few internal haemorrhagic and cystic areas arising from septum pellucidum.
On histopathological evaluation, there is presence of neurocystic and glial components. Neurocystic cells are having small round nuclei with fine stiplled chromatin and scant cytoplasm. The neurocystic cells are present in partly microcystic arrangement. Glial fibrillary component with oligodendroglia-like cells are seen. In immunohistochemistry GFAP is positive, SOX 10 is positive and Synaptophysin is negative for oligodendrocyte-like cells and positive for floating neurons so it is in favour of Myxoid glioneuronal tumor (WHO Grade 1).
These tumours feature a unique dinucleotide change at codon 385 in the platelet-derived growth factor receptor α (encoded by the PDGFRA gene), resulting in the substitution of lysine 385 into leucine or isoleucine. Molecular hallmark of MGNT is mutation in platelet-derived growth factor receptor α. The functional consequences of these mutations remain largely unexplored. This genetic variation has not been reported in any other CNS tumor.[8]
MGNTs are primarily benign with a slow-growing nature, careful monitoring is necessary after subtotal resection due local recurrence and dissemination within the ventricular system. However, even in these cases, the tumors tend to remain slow-growing and continue to be associated with a relatively favourable prognosis.[9] High-grade transformation has not been described to date.
Myxoid Glioneuronal Tumors (MGNTs) are generally considered to be low-grade, slow-growing tumors with a relatively favourable prognosis compared to high-grade gliomas. However, like any tumor, there is a potential for metastasis, though it is quite rare for MGNTs.
Intracranial metastasis: MGNTs are more likely to spread within the central nervous system (CNS) rather than outside it. This can involve cerebrospinal fluid (CSF) dissemination leading to drop metastases along the spinal cord or other areas of the brain.
Extracranial metastasis: Extremely rare, but possible, especially if the tumor undergoes malignant transformation or if there are surgical manipulations leading to dissemination.
MRI of the brain and spine: The primary modality for detecting metastatic spread. Look for new lesions or areas of enhancement that were not present on prior imaging.
CSF analysis: May be done to detect tumor cells in the CSF if there is suspicion of leptomeningeal spread.
Anatomical and radiological differential diagnosis
When considering the differential diagnosis for Myxoid Glioneuronal Tumors (MGNTs) located in the septum pellucidum, it's essential to distinguish them from other entities that may present similarly on imaging.
Colloid cyst: The most important differential diagnosis of MGNT is a colloid cyst. Colloid cysts typically present with high signal intensity on T1-weighted imaging (T1WI) and variable signal intensity on T2-weighted imaging (T2WI) in MRI. These cysts are usually attached to the anterosuperior portion of the third ventricular roof. They are rarely found at other sites, including the lateral ventricles, cerebellar parenchyma, pituitary gland, and various extra-axial locations.
Central neurocytoma: Central neurocytomas arising in the septum pellucidum characteristically demonstrate a solid-cystic appearance. These well-circumscribed masses are typically attached to the walls of the septum pellucidum within the lateral ventricles.
Oligodendroglioma: Commonly occurs in the frontal lobes. MRI features suggest calcifications common, T2 hyperintense, and often show mixed signal intensity. Enhancement varies. "Fried egg" appearance of cells on histopathology. Oligodendrogliomas and MGNTs can appear similar on imaging and may share some histopathological features, differences in cellular characteristics, genetic markers, and typical locations help in distinguishing them.
Subependymoma: Subependymoma is a rare, slow-growing tumor typically found in the fourth ventricle of middle-aged to elderly patients. When it protrudes into the lateral ventricles, it is more common than into the third ventricle. This tumor usually does not enhance on post-contrast sequences; however, it presents as a solid glial lesion with a central hyperintense signal on FLAIR imaging.
Dysembryoplastic neuroepithelial tumor: It has intracortical lesion with distinctive nodular pattern. Floating neuron are present on myxoid background. In Contrast to MGNT it exhibits BRAF or FGFR1 variants or rearrangement.
Conclusion
MGNT was classified as (Grade 1) tumor according to 5th edition of the WHO Central Nervous System Tumor Classification, 2021. Septum pellucidum is most common location. Histologically it is composed of oligodendrocyte-like cells within prominent myxoid stroma. MRI findings of MGNT are more distinctive, typically showing a cystic appearance. These cystic areas always exhibit T1- hypointensity, T2-hyperintensity images. MGNTs have been related with a dinucleotide substitution at codon 385 within the platelet-derived growth factor receptor alpha (PDGFRA) gene.
Source of Funding
Nil.
Conflict of Interest
Nil.
References
- Solomon D, Korshunov A, Sill M, Jones D, Kool M, Pfister S. Myxoid glioneuronal tumor of the septum pellucidum and lateral ventricle is defined by a recurrent PDGFRA p.K385 mutation and DNT-like methylation profile. Acta Neuropathol. 2018;136(2):339-43. [Google Scholar]
- Louis D, Perry A, Wesseling P, Brat D, Cree I, Figarella-Branger D. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. 2021;23(8):1231-51. [Google Scholar]
- Zhou J, Qu K, Lv M. A 4-year-old boy with a ventricular mass. Brain Pathol. 2022;32(5). [Google Scholar]
- Bale T, Rosenblum M. The 2021 WHO Classification of Tumours of the Central Nervous System: an update on pediatric low-grade gliomas and glioneuronal tumours. Brain Pathol. 2022;32(4). [Google Scholar]
- Caporalini C, Scagnet M, Giunti L, VC, Mei D, Conti V. Myxoid glioneuronal tumor: histopathologic, neuroradiologic, and molecular features in a single center series. Neoplasia. 2023;37. [Google Scholar]
- Lucas C, Villanueva-Meyer J, Whipple N, Bush N, Cooney T, Chang S. Myxoid glioneuronal tumor, PDGFRA p.K385-mutant: clinical, radiologic, and histopathologic features. Brain Pathol. 2020;30(3):479-94. [Google Scholar]
- Chiang J, Harreld J, Tanaka R, Li X, Wen J, Zhang C. Septal dysembryoplastic neuroepithelial tumor: a comprehensive clinical, imaging, histopathologic, and molecular analysis. Neuro Oncol. 2019;21(6):800-8. [Google Scholar]
- Brat D, Verhaak R, Aldape K, Yung W, Salama S, Cooper L. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N Engl J Med. 2015;372(26):2481-98. [Google Scholar]
- Bodi I, Curran O, Selway E, Elwes R, Burrone J, Laxton R. Two cases of multinodular and vacuolating neuronal tumour. Acta Neuropathol Commun. 2014;2. [Google Scholar]