Introduction
Pineal region tumors account for less than 1% of intracranial tumors and are very rare in occurrence.1 The World Health Organization (WHO) classification subcategorizes the pineal parenchymal tumors in to three classes: Pineocytoma (grade I), Pineoblastoma (grade IV) and Pineal parenchymal tumor of intermediate differentiation (grades II-III).2, 3 Pineoblastomas originate from pinealocytes and /or their precursors. The pinealocytes release melatonin and control the circardian cycle of sleep and wakefulness. Pineoblastoma accounts for 40% of parenchymal pineal tumors.4 It occurs more commonly in pediatric patients and as age increases the incidence decreases. Patients having pineoblastoma have poor prognosis with 5 – year survival rate <60%.4
Case Report
A 12-year-old male patient presented to SVP tertiary care hospital with complain of progressive loss of vision in both eyes since 2 months with headache and nausea. The diminution of vision was started first in left eye, followed by right eye. On examination perception of light present in both eyes with bilateral upward gaze palsy. A brain magnetic resonance imaging (MRI) revealed large ill - defined lobulated solid cystic extra axial space occupying lesion in posterior aspect of third ventricle involving pineal gland which extends up to the fourth ventricle inferiorly and compresses the tectum of midbrain with severe compression over cerebral aqueduct, possibility of pineal germinoma likely. MR venogram study was normal. Computed tomograpgy report revealed approximately 24 x 18 x 28 mm large mixed solid cystic lesion with internal specks of calcification noted in posterior aspects of third ventricle in pineal gland region. CSF cytology was negative for pathological seeding. Patient underwent ventriculoperitoneal shunt insertion surgery for obstructive hydrocephalus and sub occipital midline craniectomy for excision of tumor. Resected tumor is then sent to pathology department for Histopathological examination. Patient is followed up in neurosurgical outpatient department after 15 days.
Gross
The specimen received in formalin consist of one brownish and multiple reddish whitish tissue portions. The brownish tissue portion is measuring 1.0 cm and reddish whitish tissue portions is measuring 3.5 x 1.3 cm in aggregates.

Microscopic examination
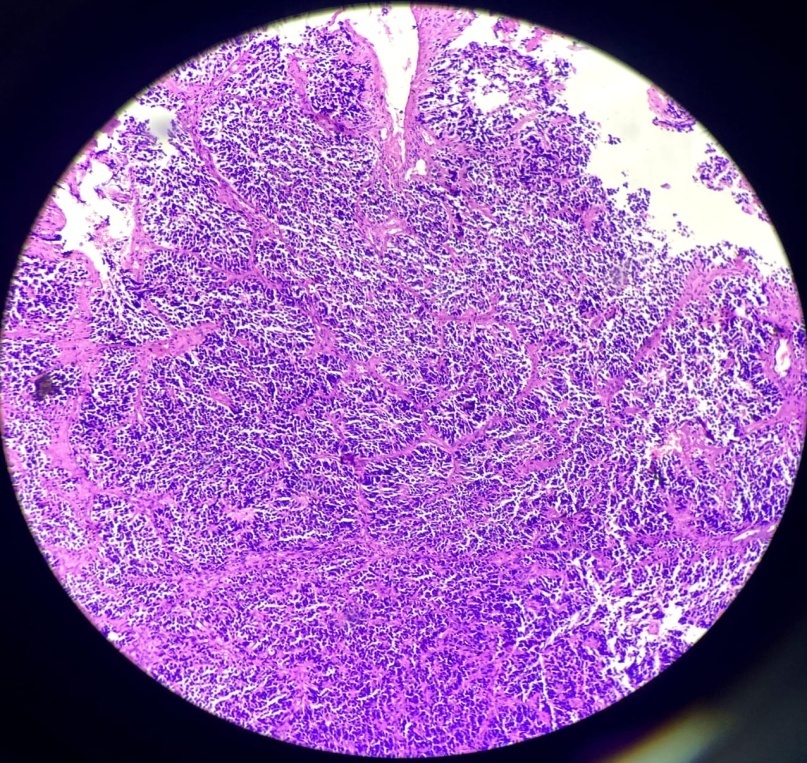
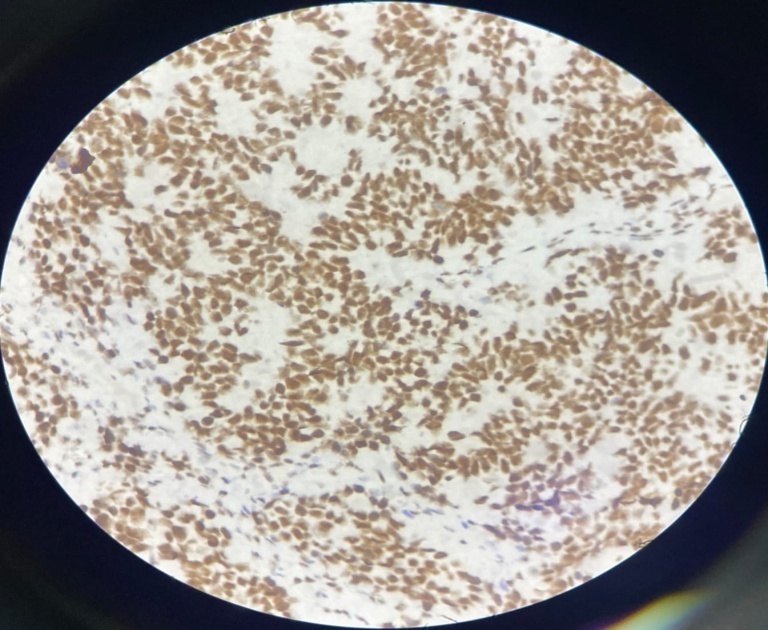
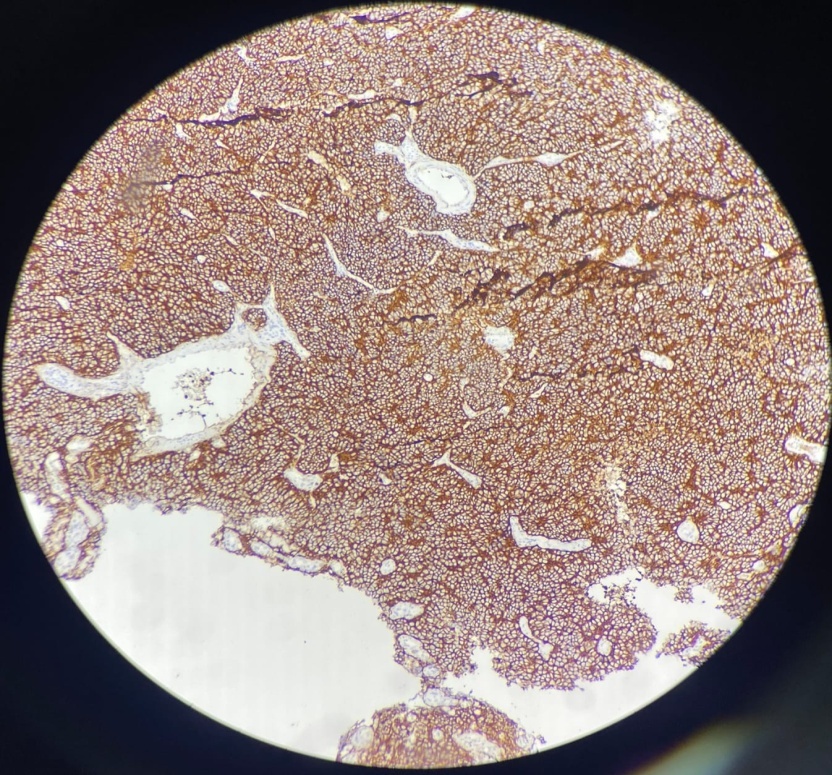
Sections show tumor tissue composed of densely packed cells arranged in nests and lobules separated by fibrous septa. The cells show high grade features having high N:C ratio with minimal cytoplasm and large hyperchromatic nuclei. Focal nuclear moulding, many atypical mitoses, brisk mitotic activity and many Homer Wright rosettes are seen and reported as Pineoblastoma, Grade IV. Immunohistochemistry was advised to confirm the diagnosis.

Discussion
Pineoblastoma is a malignant supratentorial midline primitive neuroectodermal tumor of pineal gland. Young children are more commonly affected, but can present at any age. They account for 0.001% of all primary CNS neoplasms.5 It is a WHO grade IV neoplasm with aggressive local growth and propensity for dissemination via Cerebrospinal fluid. Germline mutations of RB1 (retinoblastoma) and DICER 1 genes predispose to Pineoblastoma.6 It can occur in complex with Retinoblastoma, when latter is bilateral it is characterized as ‘trilateral retinoblastoma’.7 This tumor combination is more aggressive than an isolated pineoblastoma.8 In nonfamilial pineoblastoma, dysregulation of microRNA occurs because of mutated gene called DROSHA. Mostly patients present with complains of visual disturbances, headache and nausea. Pineoblastomas almost always associated with obstructive hydrocephalus due to compression of the cerebral aqueduct. Compression of tectal plate may also result in the Parinaud syndrome.
Radiologically, pineoblastomas are large lobulated ill-defined masses that are homogeneously hyperdense on contrast inhance computated tomography and are hypointense on magnetic resonance imaging. Grossly, pineoblastoma is a friable, soft tumor often demonstrating hemorrhage and/or necrosis. Histologically, tumor shows densely packed cells with round, hyperchromatic nuclei with focal molding. These may exhibit frequent mitotic figures, Homer Wright or Flexner- wintersteiner rosettes. Areas of necrosis may be present, reflecting the aggressive nature of tumor. Infiltration in to surrounding structures is common. In contrast to pineocytomas, pineocytomatous rosettes are not present.9 The prognosis is poor when tumor shows 7+ mitotic figures/100 HPF and necrosis. Immunohistochemistry studies are usually positive for neuronal markers such as Synaptophysin and Neuron Specific Enolase (NSE) with high Ki-67 expression.9 Electron microscopy often reveals the presence of Cytoplasmic dense core granules, short immature cell processes and occasional junctional complexes.
Pineoblastoma should be differentiated from Germinoma, Papillary tumor of pineal region, pineocytoma and metastatic carcinomas. CD-117 and SALL-4 is useful in differentiating Germinoma from Pineoblastoma. Papillary tumor of pineal region shows cells with epithelial features, intracytoplasmic vacuoles and psedopapillae and positive immunoreactivity with CAM5.2 marker.
Initial treatment for Pineoblastoma often includes a shunting procedure for obstructive hydrocephalus and removal of tumor by surgery following radiation therapy to brain and spinal cord.