Introduction
Iron is an essential divalent metal that is required for vital biological functions such as respiration, synthesis of energy, and division of cells in the body. The human body requires about 25mg/day of iron to carry out various physiological functions. However, iron absorption is only about 1–2 mg/day. This remaining iron deficit is fulfilled by macrophages that phagocytose senescent red blood cells and help recycle the iron stores. A specific hormone known as hepcidin regulates iron absorption as well as phagocytosis of senescent red blood cells thereby maintaining the normal range of total body iron and preventing both deficiency as well as a state of iron overload.1
Iron deficiency anemia is a major global health problem. According to the national health survey data, India is one of the leading countries with the highest prevalence of anemia especially in children aged between 2 years and 5 years, the most common cause being inadequate nutritional intake of iron.2 Nutritional iron deficiency is the most frequent cause of microcytic hypochromic anemia, but several other conditions such as chronic blood loss, gastrointestinal malabsorption, infections such as Helicobacter pylori, can lead to iron deficiency and anemia.3 Anemia of chronic diseases have underlying iron-restricted erythropoiesis in addition to several other mechanisms such as reduced erythropoiesis and poor response to erythropoietin being additional contributory factors. In certain hereditary conditions, the dietary iron intake may be adequate but there defects in iron absorption or its redistribution from stores. These are rare inherited iron–related anemias and are also referred to more commonly as “iron-refractory” anemias. Entities that are included in inherited iron related anemias are sideroblastic anemias- classical type and iron refractory iron deficient anemia (IRIDA). Iron-refractory iron-deficiency anemia (IRIDA) is an extremely rare genetic disorder, which is associated with absent iron absorption by the body or due to suboptimal response of the body to iron supplements, both oral and parenteral.4 This entity has been recently described and are due to mutations in the TMPRSS6 gene (mapped to chromosome 22q12-q13) which encodes Matriptase2 (MT-2).5 The prevalence of IRIDA is not exactly clear. Some studies mention the prevalence as <1 in 1,00,000,6 but it is certainly an underdiagnosed entity and should considered as a possible differential after ruling out all other known causes of iron deficiency anemia. Before dwelling further on the topic, we need to define oral iron refractoriness which is failure to respond or a suboptimal response to iron therapy given at a dose of minimum 100 mg of elemental iron per day after 4 to 6 weeks of therapy. The response is estimated by an increase in Hb of at least 1 g/dL with adequate patient compliance and after ruling out other known acquired forms of GI disorders.7
Discussion
Historical facts
IRIDA has been historically described first by Buchanan and Sheehan in 1981 in a family with 3 siblings who had initially presented as iron deficiency anemia with adequate nutritional intake and no other evidence of gastrointestinal blood loss.8 The patients did not respond to oral iron therapy and only had partial response to parenteral iron therapy. Subsequently, Brown et al in 1988 reported a family comprising of 2 sisters with history of microcytic anemias who showed failed response to oral iron therapy.9 It was Hartman and Barker in 1996, who brought to light the fact that malabsorption of iron may be the cause of microcytic anemia in two siblings following an iron challenge with 2 mg/kg of ferrous sulphate.10 Several other observers(Pearson and Lukens-1999) highlighted families having siblings who had severe microcytic anemia but showed only partial response to parenteral iron. Long term follow-up of these patients helped the observers to conclude that it was a novel gene that was most likely responsible for prevention of iron absorption by enterocytes into circulation.11 Finally, it was Finberg et al. in 2008 who classified these clinical presentations into an entity known as iron refractory iron deficiency anemia (IRIDA) along with the major abnormalities associated with mutations in the TMPRSS6 gene.12
Pathophysiology of IRIDA
Systemic iron homeostasis: Hepcidin-Ferroportin axis
Hepcidin is known to be one of the key regulators of systemic iron homeostasis which is a peptide hormone expressed by liver. The identification of hepcidin helped in deciphering and understanding how the liver centrally regulates iron homeostasis and how dysregulation of the same results in iron related disorders. Hepcidin peptide comprises of 25 amino which controls iron export to the plasma by causing induction of lysosomal degradation of ferroportin (iron exporter) in enterocytes, macrophages, and hepatocytes.13 Iron which is bound to transferrin can be thereby readily taken up by different cell types by transferrin receptor 1 (TfR1) which is ubiquitously expressed.
The peptide hormone hepcidin that is expressed by the liver, helps in maintaining iron homeostasis by regulation of iron absorption by the intestines, recycling of iron from senescent erythrocytes which is mediated by macrophages, and mobilization of hepatic iron stores. Iron export is downregulated by hepcidin by binding to ferroportin which is an iron exporter expressed on the surface of cells that release iron, accelerating its degradation thereby regulating plasma iron levels. Hepcidin is regulated by systemic availability of iron, iron requirement for erythropoiesis, hypoxia, and inflammatory conditions.14 (Figure 1)
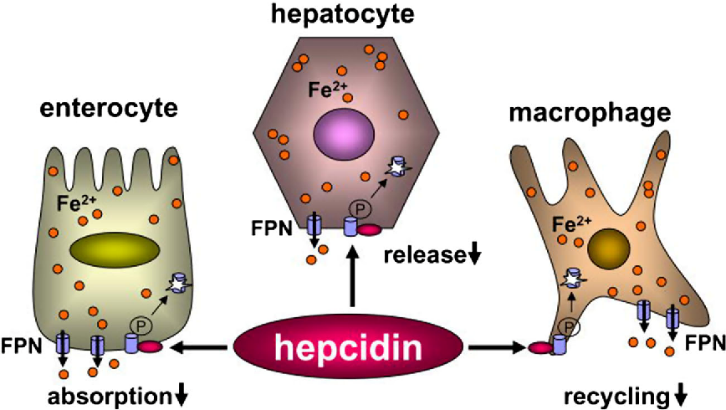
Genetic polymorphisms responsible for iron-refractory iron-deficiency anemia role of transmembrane protease serine 6 and matriptase-2
TMPRSS6 loss-of-function mutations leads to an increase in hepcidin levels that in turn is responsible for causation of iron refractory iron deficiency anemia.16 TMPRSS6 (transmembrane protease, serine 6) gene encodes matriptase-2, which is a protease that spans the membrane and is expressed by the liver primarily.17 Matriptase-2 belongs to type II transmembrane serine protease (TTSP) family, which is a group that is characterised by the amino terminus being anchored to the membrane. The protein, matriptase-2, exhibits a structure that is homologous to another transmembrane serine protease, matriptase-1. Matriptase-2 structure comprises of a large extracellular region and many structural domains, such as SEA (sea urchin sperm protein, enteropeptidase, agrin) domain, 2 CUB (C1r/C1s, urchin embryonic growth factor, bone morphogenetic protein 1) domains, 3 LDLRA (low-density lipoprotein receptor class A) domains, and a C-terminal catalytic domain that contain a triad of serine, histidine, and aspartic acid residues (Figure 2).
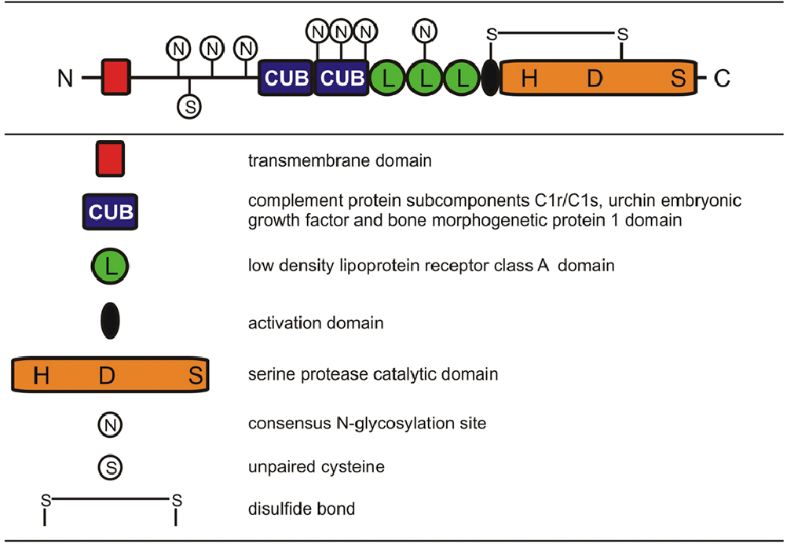
Matriptase-2 has a very crucial role to play in iron metabolism, the observations having been based on experiments carried out in various mice models that had features of IRIDA. This gene functions as a negative regulator in hepcidin production and is expressed primarily in the liver. Various studies carried out in recent times have brought to light the fact that the main role of matriptase-2 is regulating hepcidin.19, 20 Till date, numerous mutations in the TMPRSS6 gene have been detected that have been distributed in various extracellular domains in the gene. The mutations that have been reported so far in humans include approximately 20 missense mutations, 5 nonsense mutations, 10 frameshift mutations, 1 large in-frame deletion, and 9 intronic mutations. A structure of various mutations in the TMPRSS gene causative of IRIDA is depicted in Figure 3.

Matriptase-2 is synthesized as an inactive, single-chain polypeptide that is membrane bound. This inactive polypeptide undergoes multiple proteolytic cleavages during zymogen activation for conversion into active form.22
The main role that TMPRSS6 plays in iron homeostasis was first elucidated by the genetic experimentation. This was carried out in a recessive mutant mouse phenotype termed mask which was induced chemically. 23 The mask mutant was so named as it showed a progressive loss of truncal hair but had hair retained on the head. Mice with this phenotype was seen to exhibit microcytic anemia, decreased levels of plasma iron along with reduced iron stores when they were fed a standard diet for rodents in the laboratory. It was also seen that mask mice exhibited defective regulation of hepcidin regulation in response to iron deficiency and that hepcidin messenger RNA (mRNA) was elevated out of proportion in the livers of mask mutants. A genetic mapping to elucidate the underlying mutation was carried out which revealed that these mask mice had defects in splicing of TMPRSS transcript due to an underlying homozygous mutation thus eliminating the proteolytic domain of matriptase-2. The increased levels of hepcidin mRNA levels in the liver of the mask mutants proved that the main function of matriptase-2, which is a TMPRSS6 gene product, is a decrease of expression of hepcidin by the liver.
Various studies that were carried out in transgenic models and tissue culture systems have tried to explain and shed light on the mechanism of regulation of hepcidin by matriptase-2. The modulation of bone morphogenetic protein (BMP)/SMAD signaling mechanism which is a key signal transduction pathway, helps in promoting hepcidin transcription in hepatocytes (Figure 4). BMPs are ligands that are secreted and belong to the transforming growth factor beta superfamily that interact with both type 1 as well as type 2 BMP receptors at the cell membrane and activate the phosphorylation of several receptor-associated SMAD proteins (SMAD1, SMAD5, SMAD8). Once these are phosphorylated, the SMADs that are receptor-associated bind to SMAD4, form complexes that are heterodimeric, and translocate to the nucleus thus causing regulation of transcription of BMP target genes. This is done by their binding to specific elements in the promoter region.24 It has also been postulated that BMP pathway signaling gets modulated in response to iron stores in the liver. In case, there is an increase in local iron stores, the liver increases the BMP6 expression, which is a BMP ligand that plays a crucial role in promotion of transcription of hepcidin thus causing an increased expression of hepcidin which is actually an adaptive response that helps in limiting any further absorption of iron from the diet.24, 25, 26 Matriptase-2 is known to inhibit BMP signalling, and transcription of hepcidin by cleaving hemojuvelin from the cell membrane which is a glycosylphosphatidylinositol-linked protein.27 Hemojuvelin, acts as a co-receptor for BMP ligands and has a key role in promotion of signalling of hepatic BMP . This is evidenced by the fact that loss-of-function mutations in the hemojuvelin gene is responsible for juvenile hemochromatosis which is a severe form of hereditary iron overload associated with levels of hepcidin that is inappropriately low.
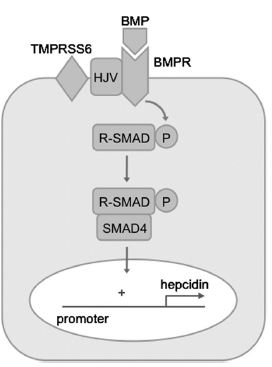
It has also been proven that genetic disruption of Tmprss6 in mice leads to low hepatic iron stores which is associated with an upregulation of hepatic expression of Bmp target genes, and also phenotypic features dependent on the coexistence of both the Bmp6 ligand and the Bmp co-receptor hemojuvelin.28, 29, 30 Together, these findings prove that increased hepcidin observed in patients with the phenotype of IRIDA phenotype is due to inability to down-regulate signaling of hepatic BMP appropriately in response to low hepatic iron stores.
IRIDA-clinical phenotype
A review of various case reports documented earlier has identified several common characteristics which were suggestive that these cases represented the spectrum of the same underlying disorder i.e. IRIDA. The key features of IRIDA include detailed clinical history including a history of compliance with oral iron therapy and a high index of clinical suspicion in patients having the following:
Congenital microcytic hypochromic anemia with Hb values ranging from 6–9 g/dL.
Very low mean corpuscular volume MCV (Range 45–65 fL)
Reduced transferrin saturation (<5%)
Defective oral iron absorption (absence of hematological improvement following oral iron therapy)
Defective utilization of iron (indicated by a partial, transient or absence of response to parenteral iron)
Autosomal recessive inheritance pattern
Other acquired causes of iron deficiency (eg, gastrointestinal blood loss) along with other causes of microcytosis both inherited and acquired (eg, thalassemia, lead toxicity) need to be excluded by extensive laboratory testing.
There is a marked overlap of clinical and hematological features of IRIDA with iron deficiency anemia. Therefore, in the presence of a lack of clinical suspicion, there is a likely possibility that the entity may be either missed altogether or under-diagnosed. Hence, a meticulous approach to testing and diagnosis is required. Subtle pointers towards the diagnosis in history and initial evaluation during the initial workup of a suspected case of IRIDA are as follows:
Mild to moderate degree of anemia with onset in infancy or early childhood.
History of anemia in other siblings or the presence of an elder sibling in the family who is being treated for chronic iron deficiency without significant clinical improvement.
Extreme Microcytosis (very low MCV) and hypochromia (low MCH) as compared to degree of anemia.
No hepatosplenomegaly and absence of stigmata of classical iron deficiency anemia such as skin and hair changes, angular cheilitis and koilonychia.
Low reticulocyte count.
Persistence of low ferritin with amelioration of anemia into adulthood.
Differential diagnosis
In most cases, the differential diagnosis of microcytic hypochromic anemia is an acquired deficiency of iron, because of either poor intake of dietary iron or due to an ongoing loss. However, various thalassemia syndromes are included in the differential diagnosis of congenital microcytic hypochromic anemias. Rarer types of congenital microcytic anemias may be seen as a result of defects in iron transport, iron uptake, and mitochondrial iron utilization.
The differential diagnosis of rarer forms of congenital microcytic anemias have been summarised in Table 1.
Table 1
Table 2
A comparison of the various causes of inherited microcytic anaemia has been summarised below
IRIDA: Studies from Indian subcontinent
Iron deficiency anemia (IDA) is a major public health problem in the Indian subcontinent. According to World Health Organization (WHO) data, the prevalence of IDA in India is 60– 70% in children and pregnant women.2
Pilot studies have been carried out by P Bhatia et al.,31 who carried out systematic screening of children which helped in the identification of children in the Indian subcontinent, who were deficient in iron and had both IRIDA phenotype as well as genotype. The study population comprised a total of 550 paediatric cases who had presented with variable degrees of anemia along with moderate to severe microcytosis. After enrolment these cases were given a trial of oral iron with assessment of response at 4–6 weeks. It was seen that 60 out of 550 cases (~11%) showed refractoriness to oral iron therapy. A further evaluation was carried out in this subset of cases to determine the frequency of IRIDA phenotype and genotype which identified IRIDA phenotype in 23 out of 60 patients. These cases with IRIDA phenotype were found to have low to very low plasma iron levels with none of the patients having normal values (mean 0.96 vs. 1.25 lmol/l). IRIDA phenotype patients had a normal range of mean plasma ferritin as compared to those without IRIDA phenotype (40.24 vs. 2.8 lg/l). The mean plasma hepcidin levels were on the higher side compared to the normal range (28.8 vs. 17.0 ng/ml).
20 out of 23 cases were subjected to sequencing (03 not done due to non-consent), results revealed, that there were a total of 65 genetic variations of TMPRSS6 in 12/20 (60%) cases which included 15 exonic and 50 intronic variations. Synonymous variations were seen in 11 out of 15 exonic variations i.e. they were non-functional without any change in amino acid sequence or silent variations. Defining the autosomal recessive nature of IRIDA, these variations were found to be either homozygous or compound heterozygous. This was one of the first and largest studies from the Indian subcontinent which highlighted the prevalence of IRIDA phenotype. This was mostly based on levels of ferritin, iron, and hepcidin. In the cohort of patients having phenotype of IRIDA, plasma ferritin levels ranged from normal (56.5%) to low-normal (43.5%). It was also seen that plasma hepcidin was high in 30% and within normal range in 70% of cases. A large proportion of cases (62%) study in the patient cohort had presented with unexplained oral iron refractoriness but did not fulfil the criteria for IRIDA phenotype.
Another study from the Indian subcontinent by Athiyarath et al., in 2015,32 5 pregnant females who had presented with anemia with failed/suboptimal response to oral iron supplements were subjected to next-generation sequencing (NGS) for 9 iron-related genes. Many rare and a few novel variants in the iron metabolism-related genes with the P555S SNP in TMPRSS6 and other potentially deleterious variations in intronic regions in TF (encoding transferrin), TRF2 (transferrin receptor 2), SLC11A2 and HAMP were found. These findings concluded that our ethnic population showed a high frequency of intronic variations predisposing to the IDA/IRIDA-like phenotype. It was also concluded by the authors in this study that these intronic variations could have a synergistic effect on the degree of anaemia in these patients.
Genome-wide association studies are needed in a large sample of our population for identification of TMPRSS6 and other iron-related gene polymorphisms and to accurately determine their frequency as iron metabolism is a very complex phenomenon. The iron homeostasis in our body is likely to be affected by the many genetic variations in both exonic and or intronic regions of genes.
Comprehensive Genomic Analysis was carried out in another large study in the Indian subcontinent in which anemia cases were systematically evaluated from 2019 to 2021. Prior to the genetic studies, patients were subjected to other screening tests (high-performance liquid chromatography, α gene sequencing, erythrocyte sedimentation rate, C-reactive protein, and tTG), followed by detection of abnormal iron profile by targeted next-generation sequencing (26-gene panel). This was supplemented with whole-exome sequencing, multiplex ligation probe amplification/mitochondrial DNA sequencing, and chromosomal microarray. Functional validation of the novel variants in ALAS2, STEAP3, and HSPA9 genes was done. The cohort comprised of 290 cases of anemia cases, out of which 41 cases (14%) were enrolled for genomic testing as per inclusion criteria. Pathogenic variants on comprehensive genomic testing were detected in a total of 23 out of 41 cases (56%). It was observed in the study after genotype-phenotype correlation that there was a significant association of frameshift/nonsense/splice variants with lesser age at presentation (0.8 months versus 9 years; P < 0.01) as compared to missense mutations. This study brought to light that a systemic evaluation could help in detecting inherited iron defects as seen in 41% (17/41) of cases, thereby suggesting, that it requires active screening and awareness programmes for detection of these rare diseases in an iron-deficient endemic population.33
Approach to diagnosis: “SAID” (structured approach to IRIDA4
A structured approach to the diagnosis of IRIDA can guide general practitioners and physicians to diagnose cases. This approach is flowchart-based based that has been suggested and is not a recommendation or a guideline and may be modified as per requirements of the case and the availability of in the health care setups4 and has been depicted below.

Genetic testing
TMPRSS 6 gene comprises of 18 exons and encodes for an 802 amino acid protein. Till date there have been more than 70 variants that have been identified throughout the TMPRSS6 gene. More than 50% of these are known to be missense variants that are responsible for an impaired enzymatic activity. De Falco et al. have also reported other variants such as nonsense mutations, insertions/deletions, and alterations in the splice site.21 The mutations that are more likely to affect protein function severely include missense mutations, frameshift and splicing mutations, etc in the serine protease domain; however, a low expression of protein or an unstable protein (reduced interaction with hemojuvelin) or a protein with partial activity have been described with mutations in the other domains. Hence, owing to the heterogeneous nature of mutations, it is of utmost importance that all 18 exons including exon-intron boundaries be screened and sequenced to identify an already described mutation, or to detect a new potentially deleterious mutation. It is also important to understand that to qualify for a confirmed diagnosis of IRIDA, the mutation should involve both the alleles as a homozygous mutation or as a compound heterozygous mutation since it is an autosomal recessive disorder.
Genetic testing is the confirmatory test for diagnosis of patients with iron deficiency having IRIDA phenotype. Individuals with clinical signs and symptoms or with suspicion of, or a positive family history of Iron-refractory Iron Deficiency Anemia should be offered genetic testing including Next-generation sequencing (NGS) test with a diagnostic sensitivity of >99%. Both massively parallel sequencing and sequencing of the entire coding region may be carried out to detect the various types of heterogenous mutations. Other molecular testing methods such as Multiplex PCR mini-sequencing assay and high-resolution melting curve analysis may be carried out for genetic testing in IRIDA.
Another algorithm for evaluating cases suspected to have IRIDA has been suggested by Heeney Finberg et al and is as follows.5 This algorithm has a major difference from the “SAID” algorithm in the inclusion of an oral iron challenge test. However, this test has become obsolete with the availability of sophisticated and more sensitive assays.

Treatment
The mainstay of treatment in IRIDA is largely about using IV /parenteral iron to have a sustained response, as it has been evidenced, that the hallmark of IRIDA is oral iron refractoriness. However, it has been observed that in few cases, response to iron therapy may not be sustained or maybe partial. Current recommendations and guidelines by Donker et al suggest an initial trial of iron in the form of ferrous sulphate (FeSo4) to be administered along with Vitamin C for 6-8 weeks before parenteral iron treatment.34 The type of TMPRSS6 gene variation determines the response to oral iron, as several mutations lead to refractoriness to iron which is almost complete. Therefore, before proceeding to parenteral iron, it is important to give a trial of oral iron. Variable degrees of response to either high dose or prolonged oral iron therapy in IRIDA cases have been noted in different studies.35, 36, 37 Acceptable hemoglobin levels along with microcytosis, low transferrin saturation, and persistent hypoferremia have been seen in few cases in these studies who were adminstered high doses (6-10 mg/kg/day) of elemental iron for almost 17 months. It was Cau M et al. who reported a case of a 5-month-old Sardinian female infant harbouring homozygous TMPRSS6 mutation38 who was initially unresponsive to oral iron and showed only partial response to IV iron. However, he noticed that the infant later showed a dramatic response to oral iron and ascorbic acid after 3 months with a rise in Hb to 12 gm/dl. It has to be ensured that treatment with oral iron and vitamin C needs to be followed up diligently along with strict patient compliance. Further requirements of IV iron therapy need to be tailor made based on the response after 8-10 weeks of trial of therapy. In many cases, therapeutic goals of acceptable hemoglobin level required for the growth and development of the child may be achieved even with partial correction of mild to moderate anemia. It has been evidenced that improvement in the degree of anemia in cases of IRIDA in children may be seen as they grow into adulthood. Screening of all other siblings of a case of IRIDA constitutes a very important aspect of treatment. Initial screening for presence and degree of anemia should be done and if present, genetic diagnosis must be offered for detection of presence of targeted mutation. Intravenous iron preparations are comprised of colloidal suspension of large iron-sucrose complexes and it is the macrophages that are responsible for phagocytosis and metabolism of these complexes before releasing iron to plasma transferrin. As compared to young patients, older patients may not require or may require only reduced iron supplementation, due to a spontaneous reduction in the requirement of iron in adulthood.39 In extremely rare cases, intravenous iron may tolerated poorly or due to poor venous access, consideration of other modalities of treatment may be warranted. In one study, addition of recombinant erythropoietin to iron therapy was done to counteract excessive deposition of iron in the liver due to the administration of large doses of parenteral iron for correction of anemia.40
Conclusion
Being a new disease entity, iron refractory iron deficiency anemia must be considered as part of diagnostic workup of microcytic anemia. The main characteristics of the disease are microcytic hypochromic anemia with low transferrin saturation and normal/high serum hepcidin levels. Incomplete or partial response to intravenous iron injections or absence of response to oral iron are important characteristic features of this disease. There is a significant overlapping of hematological features of IRIDA and classical iron deficiency anemia, mainly due to the presence of low indices in both conditions, along with a low serum iron and TSAT. Therefore, this has to be contemplated while evaluating such patients. A systematic approach to diagnostic workup is absolutely essential in patients who are being treated as iron deficiency anemia having a poor response to oral iron (iron refractoriness). A detailed history to rule out compliance issues and specialised testing for conditions such as celiac disease and H.pylori infection are unmissable. Sophisticated assays such as hepcidin assay should always be ordered for and interpreted in an appropriate clinical setting only after establishment of normal control ranges as at present no internationally standardized hepcidin assay is available. One of the most sensitive markers to identify IRIDA phenotype is Hepcidin/TSAT ratio. If this value is detected to be high, the cases should be further confirmed by ordering a genetic analysis i.e. TMPRSS6 gene analysis. Testing for the TMPRSS6 gene should include testing for all 18 exons and exon-intron boundaries of TMPRSS6 gene. The nature and type of mutations, whether homozygous or compound heterozygous should also be determined.
There are many grey zones in the diagnosis of IRIDA and therefore further studies are required to assess and interpret the levels of TSAT/hepcidin ratio in the differential diagnosis of microcytic anemia. The role of genetic factors/ epigenetic factors and other environmental factors in the clinical penetrance and pathophysiology of TMPRSS6 mutations need further analysis too. At present the optimal treatment regime is also not clearly known and requires extensive research. Owing to the rarity and complexity of the disease it is a huge challenge for the clinicians to correctly recognise and diagnose the condition thereby preventing misdiagnosis and ordering unnecessary invasive diagnostic workup and also to differentiate it from other common causes of microcytic anemia.