- Visibility 96 Views
- Downloads 26 Downloads
- DOI 10.18231/j.ijpo.2023.026
-
CrossMark

Study of hamartomatous lesions along with its fatality with review of literature
- Author Details:
-
 Prajakta D Gonjari
Prajakta D Gonjari
-
Shazia Anjum
-
Anil R Joshi
-
Bharat R Sonwane *
Introduction
The word “Hamartoma” is derived from the Greek word “hamartia,” which denotes a defect or error. Albrecht first used it to describe a deformity that resembled a developing tumour like malformation in 1904.[1] It can be described as a nonneoplastic, uni/multifocal, developmental malformation made up of a mixture of anatomically native, cytologically mature cells and tissue that exhibits an unorganised architectural pattern.[2] These tumour-like structures include birth disorder in development and inborn tissue anomalies that show themselves by excessive growth until adolescence or by the time an organ or tissue has fully developed. It may develop from any of the three germinal layers, with mesodermal-derived overgrowths being the most prevalent. They exhibit excessive proliferation of cells that are native to the organ in which they first appear, but their growth is then stopped without the ability of further expansion.[3]
Hamartomas must be distinguished from related diseases such as malformations, choristoma, teratoma and benign tumours. A choristoma (heterotopia) also known as aberrant rest, is an abnormal remnant of normal tissue. These are the growth of any histologically normal tissue found in an aberrant site.[4] Due to the potential for clonality, it might be difficult to distinguish between benign neoplasms and hamartomas. Nevertheless, a hamartoma exhibits self-limited growth in contrast to a tumour.[5] Constant dysmorphic cell proliferation that are naive to the organ in which they first appear and that also produce the cytokines required for tissue invasion but not those required for metastasis are known as benign neoplasms.[3] Teratomas are real tumours made up of tissues from each of three germ cell layers. According to the Willis, they are “real neoplasm constituted of several tissues foreign to the part in which they form”.[6]
Hamartomas are mesenchymal tumours that come in a variety of forms depending on the major tissue that makes up each one: bone forming, cartilage forming, fibre forming and non-matrix forming hamartomas. Hamartomas can develop in almost all organ systems, either randomly or in conjunction with syndromic conditions.[5]
System by system hamartomas categorised according to origin into head and neck, central nervous system, cardiothoracic, GIT, GUT, musculoskeletal.[5] Based on tissue of origin head and neck hamartomas are divided into three groups: Connective tissue, epithelial and others. There are several different types of connective tissue hamartomas, including those with vascular, lymphatic, neurologic and osseous origin.[7] This category contains cystic lymphangioma which must be distinguished from Warthin tumours, retention cyst and cystic pleomorphic adenoma.[8] Hamartomas of gastrointestinal tract can be sporadic or familial.[7]
Materials and Methods
A retrospective study was conducted in the pathology department of a tertiary health institute over a period of 2 years from July 2020 to June 2022. Total twenty cases were included in study occurring at head and neck region, chest, gastrointestinal tract and extremities. Detailed clinical history and radiological findings were obtained and taken into consideration. This is a retrospective study done on paraffin embedded tissue blocks.
Results
It was observed that twelve cases occurred in Head and neck region, one case occurred over chest, one case in Gastrointestinal tract and six cases occurred in extremities.
Hamartomas in head and neck region
12 cases were observed in head and neck region.
Laryngeal hamartoma
40 days male child admitted for inspiratory stridor within one hour of birth. Patient intubated and surfactant given. He was born at full term in uncomplicated pregnancy. Birth weight of child was 2.6 kg. Vital signs included heart rate 166/min, respiratory rate 60/min, blood pressure 90/60 mm Hg and temperature 36.5°C. Oxygen saturation in room air was 78%. Laryngoscopy revealed mass in larynx. CECT was advised to the patient. CECT revealed a compressing vascular sling around trachea and oesophagus.
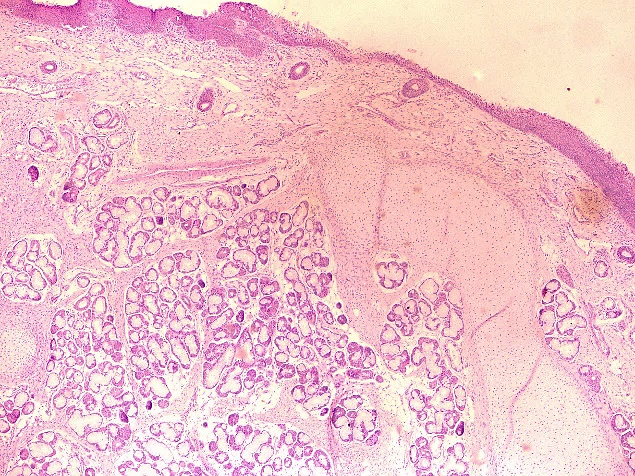
Gross specimen showed three tissue bits which were congested, firm to hard, each of size 1x0.5x0.5 cm. On cut section greyish white. Histopathology examination showed lining stratified squamous epithelium and pseudostratified columnar epithelium, underneath showed mucous gland, fibroadipose tissue, nerve bundles, cartilage tissue, congested blood vessels and focal areas of haemorrhages ([Figure 1]). Diagnosis of laryngeal hamartoma was made.
Trichofolliculoma
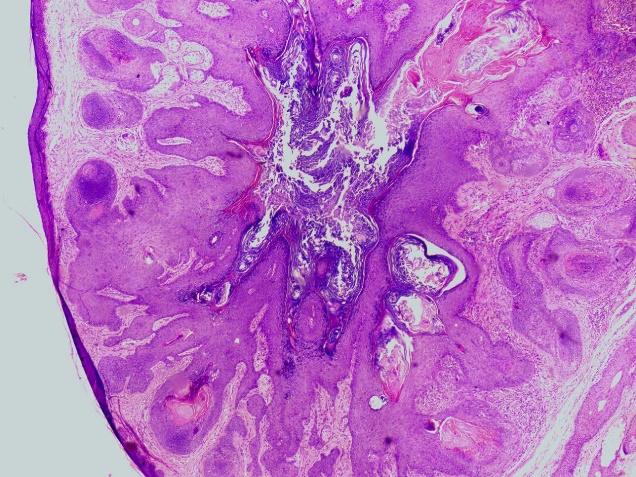
60 year male presented with swelling over left occipital region of size 1x1x0.5 cm. Swelling was firm, mobile and non-tender. No evidence of any lymphadenopathy noted. The clinical diagnosis of wart was made. Excision of nodular lesion was done. Gross examination showed nodule along with skin and attached hair of size 1.5x1.5x1 cm. Nodule of size 0.6x0.6x0.7 cm, whitish on cut section. Histopathological examination revealed lining epidermis, underneath dermis showed a cyst lined by stratified squamous epithelium and containing keratin material, fragments of hair shafts. Cyst wall surrounded by well differentiated radiating hair follicle in variable stages. At places sebaceous differentiation was noted ([Figure 2]). Multinucleated foreign body type of giant cells were noted at focal areas. Thus, the diagnosis of trichofolliculoma was made.
Other 10 cases occurred in head and neck region
Out of 10 cases, in 3 cases we received small tissue bits which on histopathological examination were found to be hamartomatous lesion and remaining 7 cases were of lymphangioma.
Clinicopathological findings are illustrated in [Table 1].
|
Case no. |
Age |
Sex |
Complaints |
Pathological findings |
|
3 |
5 year |
Male |
Swelling over right preauricular region of size 3x2x1 cm since 2 years |
Predominantly fibrous component along with adipose tissue, muscle tissue, congested blood vessels. Diagnosis- Hamartoma |
|
4 |
35 year |
Male |
Swelling over neck of size 2x1x1 cm since 2 years |
Fibroadipose and fibrocollagenous tissue, muscle tissue, congested blood vessels. Diagnosis- Hamartoma |
|
5 |
16 year |
Male |
Swelling over lower lip of size 2x2x1 cm since 5 years |
Part of salivary gland tissue, fibromuscular tissue, nerve bundles, Proliferating capillaries. Diagnosis- Hamartoma. |
|
6 |
14 year |
Female |
Swelling over left submandibular region since 2 years |
Fibro collagenous and Fibromuscular tissue along with dilated lymphatic channels. Diagnosis – Lymphangioma |
|
7 |
28 year |
Female |
Swelling over right side of neck |
Dilated lymphatic channels, fibrofatty tissue, focal lymphocytic infiltrates. Diagnosis- Lymphangioma. |
|
8 |
20 year |
Male |
Swelling on right side of neck since 1 year |
Variable sized cystic spaces with attenuated lining, scanty fluid material, Fibrocollagenous, adipose tissue, muscle bundle. Diagnosis- Cystic Hygroma (Lymphangioma) |
|
9 |
6 year |
Female |
Swelling on right side of neck |
Dilated lymphatic channels, fibrofatty tissue, focal lymphocytic infiltrates, congested capillaries. Diagnosis- Lymphangioma |
|
10 |
7 year |
Female |
Swelling over submandibular region since birth |
Dilated lymphatic channels, fibrofatty tissue, focal lymphocytic infiltrates, congested capillaries. Diagnosis- Lymphangioma. |
|
11 |
4 year |
Male |
Swelling over neck since birth |
Variable sized cystic spaces with attenuated lining, scanty fluid material, fibrocollagenous, adipose tissue and muscle bundles. Diagnosis- Cystic Hygroma (Lymphangioma). |
|
12 |
6 month |
Male |
Swelling over right side of neck since birth |
Variable sized lymphatic spaces lined by flattened cells, part of salivary gland, muscle bundles, lymphoid aggregates. Diagnosis- Cystic Hygroma (Lymphangioma). |
Hamartomas occurred over chest region
1 case were observed over chest.
Lymphangioma over chest
8 year female presented with swelling over chest since 1 year which is firm and mobile, USG suggestive of lymphangioma. Histopathological examination showed dilated lymphatics, fibrous component, muscle tissue, congested blood vessels. Diagnosis of lymphangioma was made.
Intestinal hamartoma
Neuromesenchymal hamartoma
1 case of intestinal hamartoma were observed in present study. 10 year female child was admitted with complaints of pain in abdomen since 2 days, abdominal distension since 1 day and had 1 episode of vomiting. Neither previous gastrointestinal disease nor surgical intervention was reported in past. Ultrasonography (USG) revealed dilated small bowel loops in left lumbar, left iliac fossa, periumbilical and right iliac fossa of maximum diameter 3.5 cm in right iliac fossa. Large bowel loops were collapsed and findings were suggestive of bowel obstruction. Patient taken for exploratory laparotomy. Intraoperative findings were suggestive of intraluminal growth occluding lumen spanning 4 cm in caecum and splenic flexure each along with dilated transverse colon up to 10 cm. Descending colon and sigmoid colon were collapsed. Multiple enlarged lymph nodes were noted in ileocecal region. Patient was managed by resection of ileum and colon till descending colon with ileodescending anastomosis.
Gross examination showed two loops of intestine, first loop of ileum and ascending colon of length 11 cm, on cut section multiple, small, sessile and few pedunculated polyps were noted in caecum. Second loop comprised of transverse colon with hepatic and splenic flexure of length 14 cm with one end dilated. Broad based polypoidal growth was noted near other end of size 4x1.8x1.5 cm which on cut section was soft to firm, fleshy, whitish. Microscopically mucosa was thrown into folds and supported by muscularis mucosa. Submucosa was edematous and showed prominent lymphoid aggregates. Muscle coat was hyperplastic and showed prominent nerve plexus and ganglion cells along with thick and thin-walled blood vessels ([Figure 3] a, b). Diffuse edema with lymphoid aggregates and thick and thin-walled blood vessels were noted in serosal layer. Thus, diagnosis of multiple intestinal polyps with neuromesenchymal hamartoma was made. Patient died due to cardiopulmonary arrest on next day of operation.

Hamartoma at extremity
6 hamartoma cases were observed originating in extremities.
Fibrolipomatous hamartoma of nerve
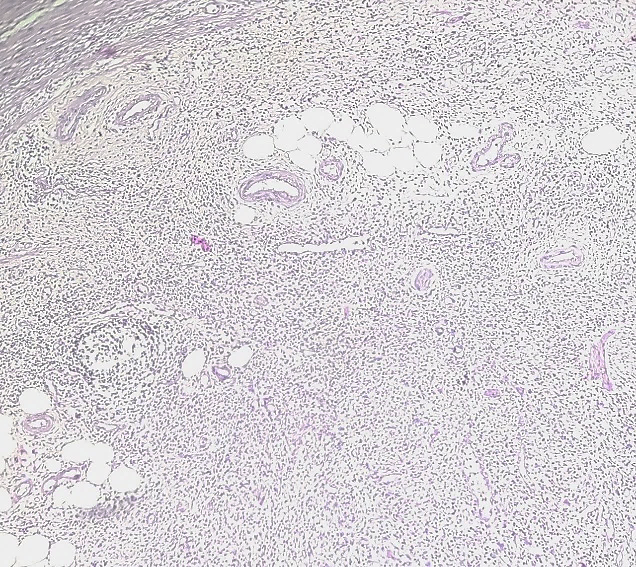
11 year female child presented with complaints of cystic swelling over left foot which was painless. Sonography report were suggestive of plexiform neurofibroma or lymphatic or vascular malformation. Excised mass on gross examination showed single capsulated, yellowish, flat, fibrofatty, soft to firm tissue piece of size 12x4.5x0.6 cm which on cut section was yellowish white. Histopathological examination revealed congested blood vessels and nerve bundles surrounded by fibroadipose tissue ([Figure 4]). Thus, the diagnosis of fibrolipomatous hamartoma of nerve was made.
Fibrous hamartoma of infancy
6 year male child presented with complaints of swelling in the left knee since birth which was painful and gradual in onset and progressively increasing in last few years. MRI revealed altered signal intensity solid lesion of size 3x2.4x2.2 cm noted in suprapatellar region deep to and on right side of quadriceps tendon which showed heterogeneously nodular enhancement with patchy areas of non-enhancement. Diagnosis of synovial sarcoma was made on MRI.
Gross examination of excised specimen showed two brownish, soft to firm tissue pieces, first of size 1.2x1x1 cm, other of size 1x1x0.5 cm, both on cut section brownish. A histopathological examination revealed interlacing bundles of spindle-shaped cells with spindle nuclei that occasionally had myxoid areas. Blood vessels with thick and thin walls as well as areas of neuronal differentiation were seen. Muscle bundles and fibrous collagenous tissue were observed. Thus, diagnosis of fibrous hamartoma of infancy was made. Within one month of excision, recurrence occurred at the site of lesion. Microscopically it showed hypertrophied muscle bundles, hypertrophied nerve bundles, fibroadipose tissue, congested blood vessels and small areas of haemorrhages. Thus, in view of previous report diagnosis of neuromuscular hamartoma was made.
For other 4 cases which occurred at extremity we received small tissue bits which on histopathological examination showed hamartomatous lesions. Clinicopathological findings of these 4 cases are illustrated in [Table 2].
|
Case |
Age |
Sex |
Complaints |
Pathological diagnosis |
|
3 |
30 year |
Male |
Swelling over forearm of size 1x0.5x0.5 cm. |
Fibroadipose tissue with thin and thick-walled blood vessels and nerve bundles. Diagnosis- Hamartoma |
|
4 |
18 year |
Male |
Swelling over right wrist of size 1x0.5x0.5 cm. |
Predominantly fibrous component along with fibroadipose tissue, nerve tissue, congested blood vessels. Diagnosis- Hamartoma. |
|
5 |
19 year |
Female |
Swelling over left lower limb of size 3x2x1 cm since 16 years |
Predominantly fibrous component along with adipose tissue, muscle tissue, thick and thin-walled blood vessels. Diagnosis- Hamartoma |
|
6 |
30 year |
Male |
Swelling over left knee of size 3x2x1 cm. |
Predominantly fibrous component along with fibroadipose tissue, muscle tissue, thick and thin-walled blood vessels. Diagnosis- Hamartoma |
Discussion
Gardner commented on the concept of hamartomas and it has been recommended that the term “hamartoma” should only be used to describe tumor-like lesions that arise during the growth of the organ or tissue in question; since these lesions are obviously non-neoplastic, the term should not be used in place of the term “benign tumour”.[9]
Review of literature of hamartoma cases illustrated in [Table 3].
|
S. No. |
Reference |
Year |
No. of cases |
Diagnosis |
|
1. |
Zapf et al[10] |
1981 |
1 |
Laryngeal hamartoma. |
|
2. |
Windfuhr et al[11] |
2004 |
26 cases review |
Laryngeal hamartoma. |
|
3. |
Ucar S et al [12] |
2019 |
1 |
Laryngeal hamartoma. |
|
4. |
Nadzirah et al[13] |
2019 |
1 |
Laryngeal hamartoma. |
|
5. |
Bharti et al[14] |
2014 |
1 |
Trichofolliculoma involving tip of nose. |
|
6. |
Choi JH et al[15] |
2019 |
1 |
Trichofolliculoma in the auricle. |
|
7. |
Lee et al[16] |
2021 |
1 |
Trichofolliculoma in the auricle. |
|
8. |
Sunil S et al[17] |
2012 |
1 |
Oral lymphangioma |
|
9. |
Kolay et al[18] |
2017 |
1 |
Oral lymphangioma |
|
10. |
Hasan et al[19] |
2022 |
17 cases review |
Lymphangioma Site- Lower lip/ buccal mucosa/ neck/ submandibular region |
|
11. |
Salas et al[20] |
1990 |
1 |
Neuromesenchymal hamartoma of small bowel. |
|
12. |
Shiomi T et al[21] |
2002 |
1 |
Neuromuscular and vascular hamartoma of the caecum. |
|
13. |
Ren B et al[22] |
2014 |
2 |
Neuromuscular and vascular hamartoma of small intestine. |
|
14. |
Sasaki et al[23] |
2020 |
1 case along with review of 26 cases |
Neuromuscular and vascular hamartoma of appendix. Sites in reviewed cases- small intestine in 25 cases and Caecum in 1 case (Shiomi et al). |
|
15. |
Silverman and Enzinger [24] |
1985 |
Review of 26 cases |
Fibrolipomatous hamartoma of nerve |
|
16. |
Roh YT et al [25] |
2020 |
2 |
Fibrolipomatous hamartoma of nerve |
|
17. |
Sotelo-Avila and Bale et al[26] |
1994 |
40 |
Fibrous hamartoma of infancy Sites- predominantly trunk followed by extremity. |
|
18. |
Vinayak et al[27] |
2011 |
1 |
Fibrous hamartoma of infancy at left thigh. |
|
19. |
Present study |
2023 |
20 |
1 case of Laryngeal hamartoma, 1 case of Trichofolliculoma, 1 case of Neuromesenchymal hamartoma of large intestine, 1 case of Fibrolipomatous hamartoma of nerve, 1 case of Fibrous hamartoma of infancy, 8 cases of Lymphangioma and 7 cases of hamartomatous lesion. |
Laryngeal hamartoma was first described by Zapf et al in 1981 in a 6-week-old child presenting with sternal retraction and stridor.[10] Systematic review by Windfuhr et al reported 26 cases of laryngeal hamartoma.[11] In our study we reported one case of laryngeal hamartoma. Hamartoma of the larynx is a relatively uncommon, non-neoplastic developmental aberration that may exhibit clinical signs of upper respiratory tract obstruction, dysphonia, stridor that worsens with time and is chronic, leading to choking and hoarseness.[12] Histologically, most laryngeal hamartomas are mesenchymal hamartomas that contain only mesodermal elements such as muscle, vascular and lymphoid tissue.[13]
Trichofolliculoma is a rare adnexal hamartomatous follicular tumour arising from hair follicle. Trichofolliculoma should be considered as a differential for a nodular mass having central pitted area containing hair and with villi around the central cyst.[16] The lesion mimics dermal nevus, basal cell carcinoma, epidermoid cyst and trichoepithelioma.[14] Treatment is typically not required because trichofolliculoma is frequently asymptomatic. However, because trichofolliculoma might result in deformity, surgical excision is the preferred choice of treatment.[15], [16] In our study we reported one case of trichofolliculoma.
Lymphangiomas are benign hamartomatous malformation arising from sequestration of lymphatic tissue.[3], [17] Three types of lymphangioma have been described: Lymphangioma simplex, Cavernous lymphangioma and Cystic lymphangioma (Cystic hygroma).[18] Hasan et al reported a case of lymphangioma of the lower lip and also reviewed 16 more cases of lymphangioma of lower lip, buccal mucosa, neck and submandibular region.[19] In our study, 7 cases of lymphangioma were found in head and neck region and 1 case occurred over chest. Surgical excision is the treatment of choice.
Neuromesenchymal hamartoma is a very rare gastrointestinal tract hamartomatous lesion. Neuromesenchymal hamartoma were first described by Fernando and McGovern in 1982.[22] Neuromesenchymal hamartoma is more appropriate term to replace neuromesenchymal and vascular hamartoma as proposed by Salas A et al.[20] In our case we noted multiple intestinal polyps with neuromesenchymal hamartoma of large bowel. Shiomi et al[21] reported one cases in which origin of hamartoma is from caecum. Sasaki et al[23] reported a case of Neuromuscular and vascular hamartoma in appendix and also reviewed 26 cases of neuromuscular and vascular hamartoma of which 25 cases were of small intestine and one case of caecum. In our case since patient died on next day of operation therefore genetic work up could not be done. Different types of hamartomatous polyps may be distinguished, such as Peutz-Jeghers syndrome, Cowden syndrome and hamartomatous polyp of Crohkhite-Canada type.[28]
Fibrolipomatous hamartoma is rare condition in which normal forms of adipose and fibrous tissue diffusely infiltrate the peripheral nerve.[29] It is rare condition that commonly affects the nerves of upper extremities, particularly the median nerve.[30] Silverman and Enzinger reported 26 cases of upper and lower extremity fibrolipomatous hamartoma.[24] We reported case of lower extremity fibrolipomatous hamartoma. This condition occurs relatively at young age. On average, 71% of reported cases presented with fibrolipomatous hamartoma before the age of 30 years.[25] During the earliest stage clinical symptoms were typically absent; however after several years of gradual and progressive growth, neuronal compression symptoms such as tenderness, decreased sensation and paresthesias begin to develop.[31]
Fibrous hamartoma of infancy was first described by Reye in 1954. Reye believed they represented a reactive phenomenon since they demonstrated the orderly and progressive growth of adult fibrous tissue.[32] Enzinger studied 30 cases and coined the term fibrous hamartoma of infancy.[33] In about 20% of cases, fibrous hamartoma of infancy manifest as congenital lesion. It is usually occurs in children 2 years of age.[34] However, cases occurring in older children are well documented.[35] Study conducted by Sotelo-Avila and Bale et al. found that predominant site for fibrous hamartoma of infancy is trunk followed by extremity.[26] In our case we observed the lesion at left knee. Local recurrence is rare and treatment is largely successful by local excision. The clinical course is usually benign and prognosis is excellent.[36] We observed fibrous hamartoma of infancy in a 6-year male child with recurrence within one month of excision. According to the study by Enzinger, the local recurrence rate is about 16%, which could be explained by insufficient excision of lesion.[33] The differential diagnosis for subcutaneous swelling in an infant includes both benign and malignant soft tissue tumours such as epidermoid cyst, recurring digital fibrous tumour, juvenile aponeurotic fibroma, juvenile hyaline fibromatosis, palmoplantar fibromatosis, histiocytoma, leiomyosarcoma, fibrosarcoma.[27], [37]
In our study we reported total 20 hamartoma cases, out of which patient with intestinal hamartoma died due to cardiopulmonary arrest. Laryngeal hamartoma patient presented with stridor which is near to fatal. Recurrence was noted in a case of fibrous hamartoma of infancy. All other patients post operatively were clinically stable.
Conclusion
It is important to differentiate hamartomas from related lesion like choristomas, teratomas and benign tumours to avoid aggressive treatment and morbidity. Hamartomas are usually not associated with significant morbidity but for the size and location of tumour. Hamartomas can affect virtually any system. Clinical course of hamartoma is mostly benign, having no tendency to regress spontaneously. There are chances of recurrence, especially when excised incompletely. There are no any other documented treatment options for hamartoma and close clinical follow up is required with molecular studies in syndromic types of hamartoma.
Source of Funding
None.
Conflict of Interest
None.
References
- WB Ober. Selected items from the history of pathology: Eugen Albrecht, MD (1872-1908): hamartoma and choristoma. Am J Pathol 1978. [Google Scholar]
- S Patil, R S Rao, B Majumdar. Hamartomas of the oral cavity. J Int Soc Prev Community Dent 2015. [Google Scholar]
- E Singh, VK Varsha. Hamartomas of the head and neck: A case report and review. J Int Res Med Pharm Sci 2016. [Google Scholar]
- R Bansal. Choristomas of the oral cavity. Indian J Pathol Microbiol 2010. [Google Scholar]
- FL Herrán, CS Restrepo, DIA Gómez, T Suby-Long, D Ocazionez, D Vargas. Hamartomas from head to toe: an imaging overview. Br J Radiol 1071. [Google Scholar]
- S Acharya, A Tayaar, S Adirajaiah, G Kulandswamy. Oral Teratoma with a Primitive Neuroectodermal Tumor Component: A Case Report. Int J Clin Pediatr Dent 2011. [Google Scholar]
- . Hamartomas of Body: A Revisited Entity - An Experience of a Tertiary Care Hospital. Med J Dr. D.Y. Patil Vidyapeeth 2022. [Google Scholar]
- M Fukunaga. Expression of D2-40 in lymphatic endothelium of normal tissues and in vascular tumours. Histopathology 2005. [Google Scholar]
- CE Tomich, DG Gardner. The concept of hamartomas: Its relevance to the pathogenesis of odontogenic lesions. Oral Surg Oral Med Oral Pathol 1978. [Google Scholar]
- S Hasan, SA Ahmad, M Kaur, R Panigrahi, S Panda. Lymphangioma of the Lower Lip—A Diagnostic Dilemma: Report of a Rare Case with a Brief Literature Review. Case Rep Dent 2022. [Google Scholar] [Crossref]
- TA Silverman, FM Enzinger. Fibrolipomatous hamartoma of nerve. A clinicopathologic analysis of 26 cases. Am J Surg Pathol 1985. [Google Scholar]
- T Shiomi, K Kameyama, Y Kawano, Y Shimizu, T Takabayashi, Y Okada. Neuromuscular and vascular hamartoma of the cecum. Virchows Arch 2002. [Google Scholar]
- C Sotelo-Avila, PM Bale. Subdermal fibrous hamartoma of infancy: pathology of 40 cases and differential diagnosis. Pediatr Pathol 1994. [Google Scholar]
- RS Vinayak, S Kumar, S Chandana, P Trivedi. Fibrous hamartoma of infancy. Indian Dermatol Online J 2011. [Google Scholar]
- B Zapf, WB Lehmann, GG Snyder. Hamartoma of the larynx: an unusual cause for stridor in an infant. Otolaryngol Head Neck Surg 1981. [Google Scholar]
- JP Windfuhr. Laryngeal hamartoma. Acta Otolaryngol (Stockh) 2004. [Google Scholar]
- Ş Uçar, P Zorlu, I Yıldırım, Ö Metin. Hamartoma of the Larynx: An Unusual Cause of Stridor. Balk Med J 2014. [Google Scholar]
- N Dani, CY Liu, YP Wong, BS Goh. Laryngeal Hamartoma Presenting as Congenital Stridor in Neonate: A Rare Entity. Acad J Ped Neonatol 2019. [Google Scholar]
- JN Bharti, B Dey, P Gautam, P Desai, V Kamal. Trichofolliculoma Presenting as Lobulated Mass: A Rare Presentation. Int J Trichol 2014. [Google Scholar]
- A Salas, F Casellas, J Sanz, F Garcia, C Margarit, JR Malagelada. Neuromesenchymal hamartoma of the small bowel. J Clin Gastroenterol 1990. [Google Scholar]
- T Sasaki, T Furuhata, M Nishimura, T Ono, A Noda, H Koizumi. An extremely rare case of neuromuscular and vascular hamartoma of the appendix. Surg Case Rep 2020. [Google Scholar]
- SK Kolay, R Parwani, S Wanjari, P Singhal. Oral lymphangiomas - clinical and histopathological relations: An immunohistochemically analyzed case series of varied clinical presentations. J Oral Maxillofac Pathol JOMFP 2018. [Google Scholar]
- S Sunil, D Gopakumar, BS Sreenivasan. Oral lymphangioma - Case reports and review of literature. Contemp Clin Dent 2012. [Google Scholar]
- HY Lee, EK Kim, HS Choi, J Jeong. Trichofolliculoma in the Auricle. Ear Nose Throat J 2021. [Google Scholar]
- B Ren, W Cao. Neuromuscular and vascular hamartoma: is it a true hamartoma?. J Clin Pathol 2014. [Google Scholar]
- JH Choi. Umbilicated Hairy Auricular Mass Mimicking Accessory Tragus. J Audiol Otol 2020. [Google Scholar]
- YT Roh, SW Song, C Jeong, Y Kang, IJ Park. Carpal Tunnel Syndrome Caused by Lipofibromatous Hamartoma of the Median Nerve. J Korean Neurosurg Soc 2020. [Google Scholar]
- E Cauchin, Y Touchefeu, T Matysiak-Budnik. Hamartomatous Tumors in the Gastrointestinal Tract. Gastrointest Tumors 2015. [Google Scholar]
- Y Tahiri, L Xu, J Kanevsky, M Luc. Lipofibromatous hamartoma of the median nerve: a comprehensive review and systematic approach to evaluation, diagnosis, and treatment. J Hand Surg 2013. [Google Scholar]
- S Agarwal, SC Haase. Lipofibromatous hamartoma of the median nerve. J Hand Surg 2013. [Google Scholar]
- JA Plaza, DV Kazakov, G Casas, L Requena, O Sanmartin, D Kacerovska. Fibrolipomatous hamartoma of the nerve: A clinicopathologic report of 13 cases. J Am Acad Dermatol 2014. [Google Scholar]
- RD Reye. A consideration of certain subdermal fibromatous tumours of infancy. J Pathol Bacteriol 1956. [Google Scholar]
- FM Enzinger. Fibrous Hamartoma of Infancy. Cancer 1965. [Google Scholar]
- GE Dickey, C Sotelo-Avila. Fibrous hamartoma of infancy: current review. Pediatr Dev Pathol 1999. [Google Scholar]
- CD Fletcher, G Powell, SV Noorden, PH McKee. Fibrous hamartoma of infancy: a histochemical and immunohistochemical study. Histopathology 1988. [Google Scholar]
- E Carretto, P Igna, R Alaggio, F Siracusa, C Granata, A Ferrari. Fibrous hamartoma of infancy: an Italian multi-institutional experience. J Am Acad Dermatol 2006. [Google Scholar]
- J Albukerk, H Wexler, M Dana, J Silverman. A case of fibrous hamartoma of infancy. J Pediatr Surg 1979. [Google Scholar]