Introduction
Primary pigmented nodular adrenocortical disease (PPNAD) is rare and manifests as ACTH-independent Cushing's syndrome (CS). Bilateral adrenocortical hyperplasia- multiple small cortical pigmented nodules mostly <1cm in diameter. Majority cases are diagnosed in second or third decade of life. Presentation of PPNAD in early childhood is very rare. Familial and sporadic cases reported are associated with germline inactivating mutations of the PRKAR1A gene. Association with Carney complex (skin lentigines, myxomas and other non-endocrine and endocrine tumors). PPNAD has a characteristic gross and microscopic appearance.
Case Report
We hereby present a report of three cases of PPNAD with unusual histology. The age at presentation ranged from one to four years. Bilateral adrenalectomy was performed in all patients.
Table 1
Patient details with signs and symptoms
All patients had ACTH independent cushing syndrome [CS] with high cortisol level which was cyclical in one case. Two patients showed spotty pigmentation which is one of the signs of Carney’s complex.
Table 2
Hormonaltest
Table 3
Family history and genetic studies
|
|
Case 1 |
Case 2 |
Case 3 |
|
Genetic studies |
Results awaited |
Not done |
Positive for inactivating PRKAR1A gene mutation |
|
Family history |
Not Contributory |
Not Contributory |
Not Contributory |
Diagnosis of PPNAD was made on typical clinical and laboratory features. In one case, PRKAR1A mutation was confirmed.
Gross examination
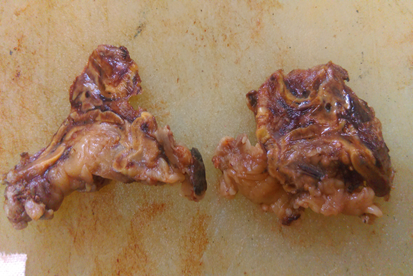
Bilateral adrenal Glands are removed in all cases. External and cut section is grey brown in colour. Nodules are not appreciated in external and on cut section (Figure 1, Figure 2, Figure 3).


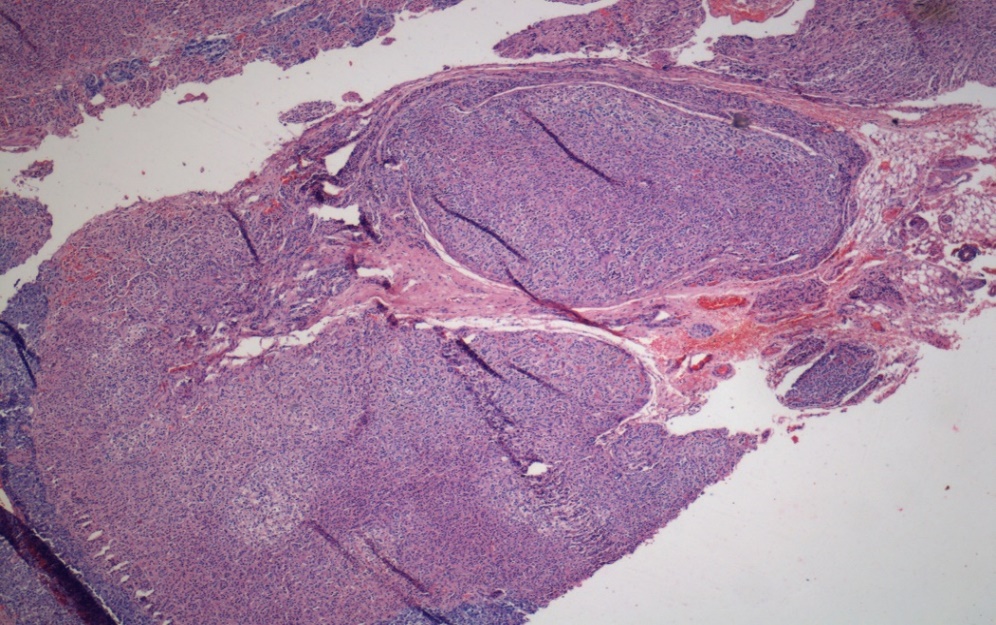
Microscopy
Heamtoxylline and eosin{H&E} stained slide (Figure 4, Figure 5) showed unencapsculated nodules composed of large eosinophillic polygonal cells with bland nuclei and clear vacuolated lipid rich cytoplasm. The characteristic liopfuschin pigment is not present in these cases and this is the unusual histology in these cases.
Figure 4
[H & E 10 X] Adrenal cortex - unencapsculated nodules beneath and abutting capsule composed of clear vacuolated lipid rich cytoplasm with bland nuclei. The Characteristic lipofuschin pigment is not seen
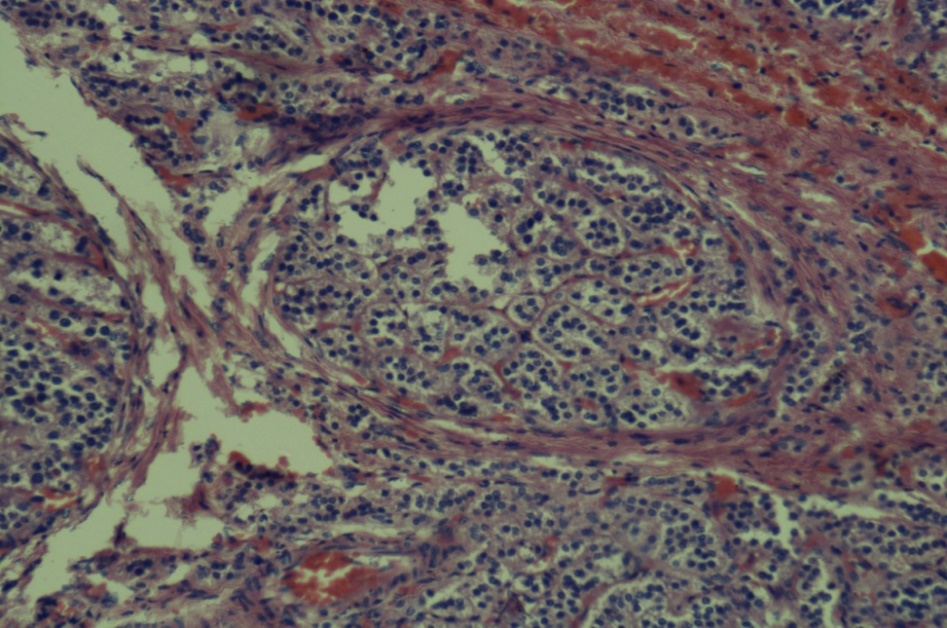
Reticulin staining pattern can be used for differential diagnosis of endocrine gland lesion in adrenal gland. Short thick, anastomosing fibers and nodular reticulin staining pattern is seen in adenoma as seen in Figure 6.

Immunohistochemistry study done with synaptophysin, marker of neuroendocrine cells which showed positivity in cortical nodules (Figure 7)
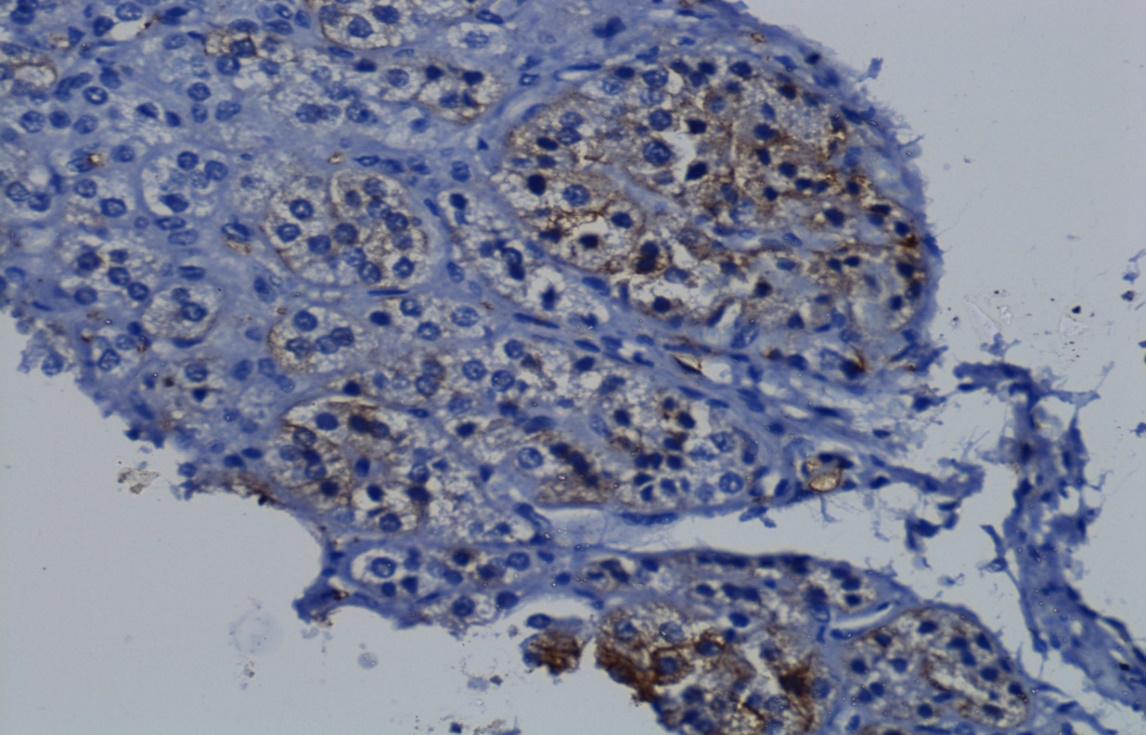
Discussion
PPNAD is a rare but an important cause of hypercortisolism especially in the pediatric age group with characteristic gross multiple small and pigmented nodules, and microscopic features such as synaptophysin positive, lipofuscin pigmented nodules, atrophic cortex in between nodules of the adrenal glands. Despite the absence of these characteristic pigmentation, these cases labelled as PPNAD because of Positive Liddle's test, synaptophysin positivity, association with Carney complex, Inactivating mutations of the PRKAR1A gene. The aim of this report is to highlight these atypical histologic features in cases of PPNAD in pediatric age group.
Most subtle pathological manifestations are seen in the young patient. The most dramatic and florid involvement was seen in the oldest patient. As the age advances, the amount of pigment lipofuschin increases. Hallmark pigmentation in the nodules seen in adult patients may not be appreciated at such an early age of presentation as seen in our 3 cases.
Cushing syndrome in the pediatric age group is a rare entity and most of the time there is a significant delay in diagnosis. Cushing syndrome secondary to PPNAD can present with typical or atypical features, but there are a number of clinical features that can help towards the diagnosis.
The symptoms are often mild and patients usually present much after the onset of symptoms.1 The disease usually presents in the second decade, but an age range of 4–44 years has been recorded in the literature.1, 2 There is a female preponderance.3
Pathologically, most nodules are small in size (usually <4 mm) and surrounded by atrophic cortex. Cells are usually large with clear or eosinophilic cytoplasm with many of them containing large amountsof coarse brown lipofuscin pigments. They have high Synaptophysin expression and it is suggestive of its neuro-endocrine nature.
The treatment of choice for PPNAD is bilateral adrenalectomy. Unilateral or subtotal adrenalectomy was followed by recurrence.2 Bilateral adrenalectomy is indicated to avoid the morbidity associated with CS. PPNAD does not increase the risk for malignancy.4 The earlier terms used to describe this condition include primary micronodular adrenal disease, micronodular adrenal hyperplasia, pigmented adrenal micronodular dysplasia and primary adrenocortical micronodular adenomatosis.3, 5 The term PPNAD was first used by Carney in 1984 and later supported by Travis et al.2, 6
Carney Complex is an autosomal dominant multiple neoplasia characterised by skin pigmentation, multiple endocrine neoplasia (PPNAD, thyroid adenoma or tumor, sertoli cell calcification, ovarian cyst, and/or growth hormone secreting pituitary adenoma), nonendocrine tumors including atrial myxoma, cutaneous myxoma, osteochondromyxoma.
Initial biochemical evaluation should be carried out the same way as evaluating any other patients with Cushing syndrome to confirm excessive cortisol. The second step is to establish whether it is ACTH dependent or ACTH independent. A low ACTH level with high cortisol level in a patient with Cushing syndrome is suggestive of ACTH independent of Cushing syndrome as seen in our study cases. Sequential low-dose-high-dose dexamethasone suppression test (Liddle’s test) can be useful to differentiate PPNAD from other primary adrenocortical lesions. The test consists of dexamethasone administration at the dosage of 0.5 mg every six hours for 48 hours and 2 mg every six hours for another 48 hours and collection of 24 hours of urine for determination of cortisol excretion baseline and every 2 days. Paradoxical rise in urinary cortisol excretion of more than 50% is indicative of PPNAD.
The pathogenesis of PPNAD has recently been well-established. Sasao et al. observed that nodules of PPNAD arise from the zona reticularis and demonstrate autonomous hypersecretion of cortisol, supporting the theory of abnormal development of the zona reticularis.7 Recently, molecular studies have demonstrated inactivating mutations of the PRKAR1A gene on chromosome 17q22-23 or of the PDE11A gene, both of which are key components of the cAMP signaling pathway.4